No dia 8 de maio foi celebrado o Dia Internacional da Talassemia, doença hereditária que se manifesta de forma bastante variada, entre casos mais leves (talassemia minor) e mais graves (talassemia major).
Embora seja uma doença rara, o diagnóstico precoce da talassemia major é fundamental para que possamos iniciar o tratamento rapidamente e evitar complicações. A Organização Mundial da Saúde estima que cerca de 60.000 crianças com talassemia grave nascem a cada ano e estima-se que no Brasil haja em torno de 1.000 pacientes com a forma grave.
Mas o que é a talassemia?
A talassemia é uma doença hereditária, causada por uma mutação genética que leva a uma redução da produção da hemoglobina, resultando em um quadro de anemia de graus variados. É uma doença autossômica recessiva, ou seja, ambos os pais devem ser afetados ou portadores para que haja a manifestação em seus descendentes. A ocorrência da talassemia major ocorre quando a criança herda a mutação recessiva de ambos os pais, conforme pode ser observado na figura 1.
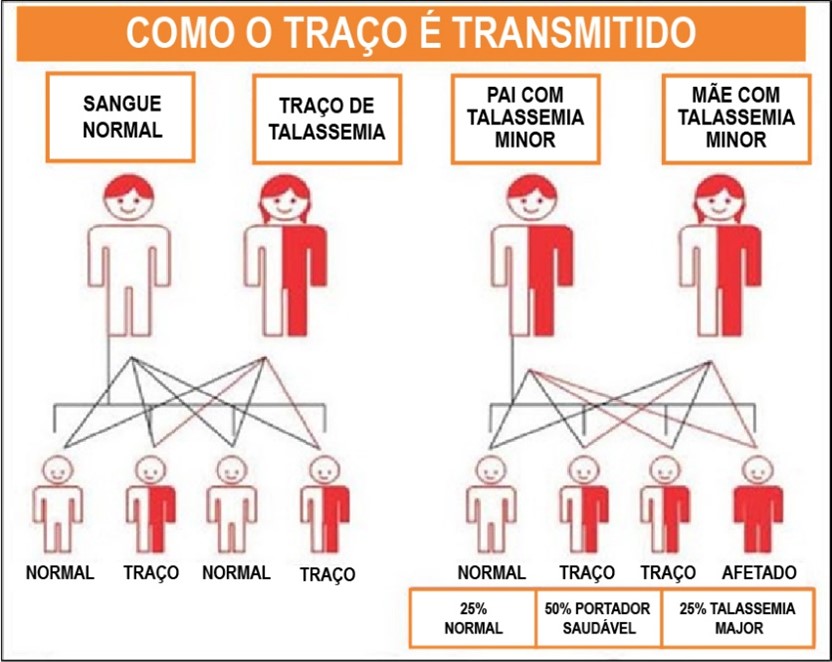
Figura 1: Herança autossômica recessiva.
Fonte: TIF.
Mas o que é a hemoglobina?
A hemoglobina (Hb) é uma proteína, que fica dentro das nossas células vermelhas do sangue (hemácias), responsável pelo transporte de oxigênio em nosso corpo, desde os pulmões até todos os tecidos do organismo. A hemoglobina (figura 2) é constituída em grande parte pela HbA, que possui duas cadeias α e duas cadeias β e, caso ocorram mutações genéticas que causam alterações na produção dessas cadeias, gera uma produção inadequada de glóbulos vermelhos, o que chamamos de eritropoese ineficaz. Com isso, a depender do tipo de talassemia, pode ocorrer a necessidade de transfusões regulares de sangue desde os primeiros meses de vida da criança.
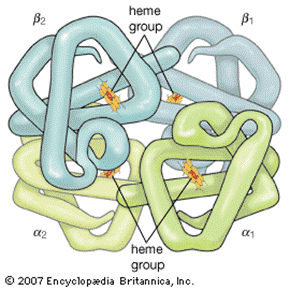
Figura 2: Estrutura da hemoglobina, constituída por duas cadeias alfa e duas cadeias beta.
Existem dois tipos de talassemia:
– Talassemia α: causada por alterações genéticas no cromossomo 16, que resultam em redução ou ausência das cadeias α-globina, levando a um acúmulo das cadeias β restantes.
Existem quatro formas de manifestação da talassemia alfa: portador silencioso, traço alfatalassemia, doença da hemoglobina H e hidropsia fetal, esta última incompatível com a vida.
– Talassemia β: causada por alterações genéticas no cromossomo 11, levando a uma redução da produção das cadeias β. A talassemia beta é também chamada de “Anemia do Mediterrâneo”.
Existem três formas de manifestação da talassemia beta: talassemia minor ou traço talassêmico beta, talassemia intermediária e talassemia major.
E quais os sintomas?
As manifestações clínicas irão depender do tipo de talassemia e a sua forma de apresentação é, portanto, bastante heterogênea, variando desde quadros assintomáticos até casos de anemia severa.
Nos casos de talassemia major, as manifestações clínicas podem se iniciar logo nos primeiros meses de vida, com sinais como: palidez da pele e mucosas, coloração amarelada da pele (icterícia), aumento do baço, deformidade dos ossos da face e ossos. Nos casos de talassemia minor, geralmente observamos alterações no hemograma como anemia microcítica hipocrômica (glóbulos vermelhos menores e mais pálidos que o habitual), sem manifestações clínicas significativas, sendo por vezes observada palidez (figura 3).
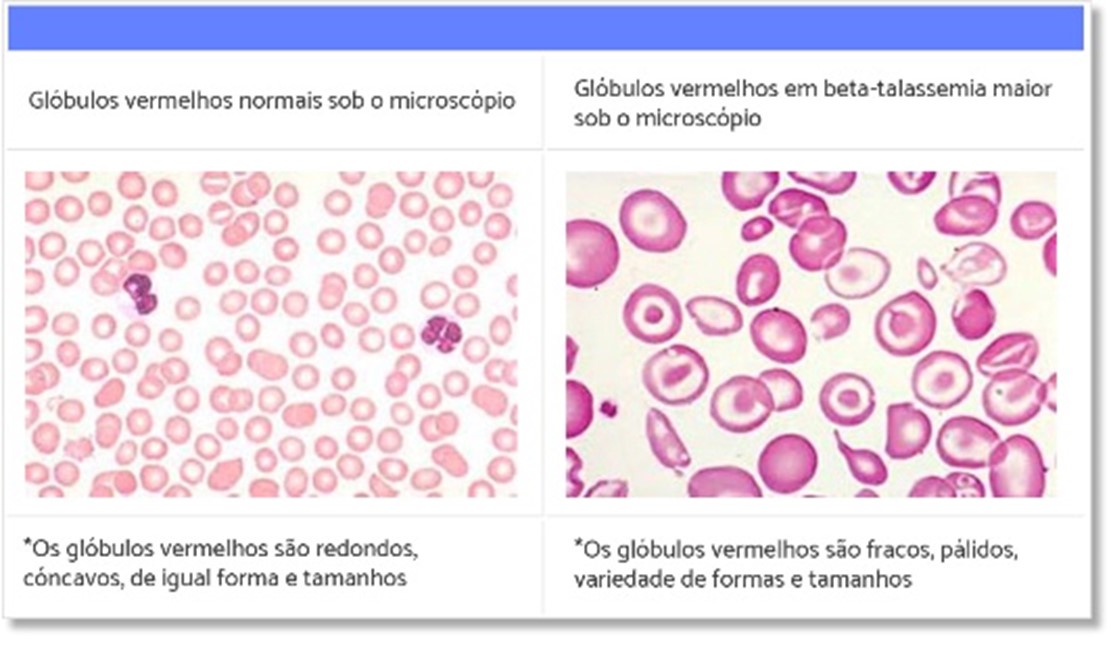
Figura 3.
Fonte: ABRASTA.
O diagnóstico das talassemias é suspeitado pela história clínica detalhada, avaliação criteriosa da história familiar e exame físico do paciente. Os exames laboratoriais fundamentais são: hemograma, reticulócitos, eletroforese de hemoglobina (talassemia beta) e testes genéticos. A talassemia alfa pode também ser suspeitada em alguns casos no teste do pezinho, quando houver detecção da Hemoglobina de Barts.
O tratamento também varia de acordo com o tipo e a gravidade da doença, sendo nos casos de talassemia major necessária a transfusão de sangue. Complicações, como a sobrecarga de ferro, são tratadas com medicamentos que eliminam o excesso de ferro do organismo, chamados de quelantes de ferro.
O tratamento curativo é o transplante de medula óssea; além deste existem novos tratamentos com excelentes resultados já realizados fora do Brasil, como a terapia gênica, atualmente realizados em casos específicos. Esperamos que em breve estejam acessíveis para um maior número de pacientes.
O tratamento da talassemia é baseado em protocolos estabelecidos pelo Ministério da Saúde. No site da ABRASTA é possível localizar os centros de tratamento especializados no Brasil.
Aconselhamento genético
É fundamental que todo paciente com talassemia receba aconselhamento genético e acolhimento de seu médico. Os pacientes portadores do traço talassêmico devem ser corretamente orientados a respeito da hereditariedade da sua condição, para que possam fazer escolhas reprodutivas conscientes e baseadas em evidências científicas.
Saiba mais:
https://bvsms.saude.gov.br/bvs/publicacoes/orientacoes_diagnostico_tratamento_talassemias_beta.pdf
Federação Internacional de Talassemia. https://thalassaemia.org.cy/
Hematologia e Hemoterapia Pediátrica: Um Guia Prático. 1. ed. Rio de Janeiro: Atheneu, 2022. Série Atualizações Pediátricas SPSP.
Relatora:
Ana Claudia Carramaschi V. Soares
Vice-Presidente do Departamento Científico de Hematologia e Hemoterapia da Sociedade de Pediatria de São Paulo

